

What are Biotech Algorithms?
Biotech Algorithms are a set of computational rules that analyze and interpret complex biological data. They typically mimic natural processes or they use machine learning to design things like new molecules or DNA structures.
BLAST:
Basic Local Alignment Search Tool is an algorithm that finds similarities between control sequences and sequences used within an experiment. It does this by searching within its own database to try and find a match.

How BLAST works:
- Seed Finding: The first step of the process requires breaking the query sequence apart into fixed-length words like 3 amino acids or 11 nucleotides. Then it searches its own databases for exact or super similar sequence matches.
- Extension: After it finds promising matches, it extends these matches in the upward direction and the downward direction. Next, it calculates the initial alignment scores without any gaps.
- Gapped Alignment: Finally, the algorithm begins to introduce gaps to simulate insertion and deletion. This allows the alignment score to increase in its accuracy. The E-score shows the statistical significance of the score, the lower the better.
Common BLAST variants:
- blastn: This variant compares a nucleotide sequence to a database of nucleotide sequences. It uses the forward sequence and the complement backward sequence.
- tblastx: This variant uses amino acids. tblastx converts a nucleotide sequence into an amino acid sequence with all six reading frame possibilities. It then does the same thing to the nucleotides in the database to compare the sequences in 36 directions.
Needleman-Wunsch:
The Needleman-Wunsch technique was created in 1970. It lines up two sequences from the beginning to the end to find the best overall alignment between the two of them.
Global Alignment: This aligns the entire sequence. It also makes sure everything is considered even if there is low similarity over some regions.
How Needleman-Wunsch Works?
- Initialization: A scoring matrix is created with the dimensions of sequence 1 with one extra and the dimensions of sequence 2 with one extra. The first row and first column are filled with gap penalty values, which represent the cost of aligning characters with gaps. This step establishes the base conditions for the alignment.
- Matrix Filling: Each remaining cell in the matrix is filled by calculating the best possible score from three options: aligning the current characters from both sequences, inserting a gap in sequence 1, or inserting a gap in sequence 2. The highest of these three scores is selected for each cell to achieve the best accuracy.
- Traceback: After the matrix is full, the best alignment is determined by tracing the matrix back from the bottom up. This allows for the algorithm to determine where there are matches, mismatches, and gaps in the final alignment.
AlphaFold:
AlphaFold is an AI tool that predicts a protein’s 3D structure based on its amino acids sequence with high accuracy. It also predicts interactions between RNA, DNA, and other proteins.
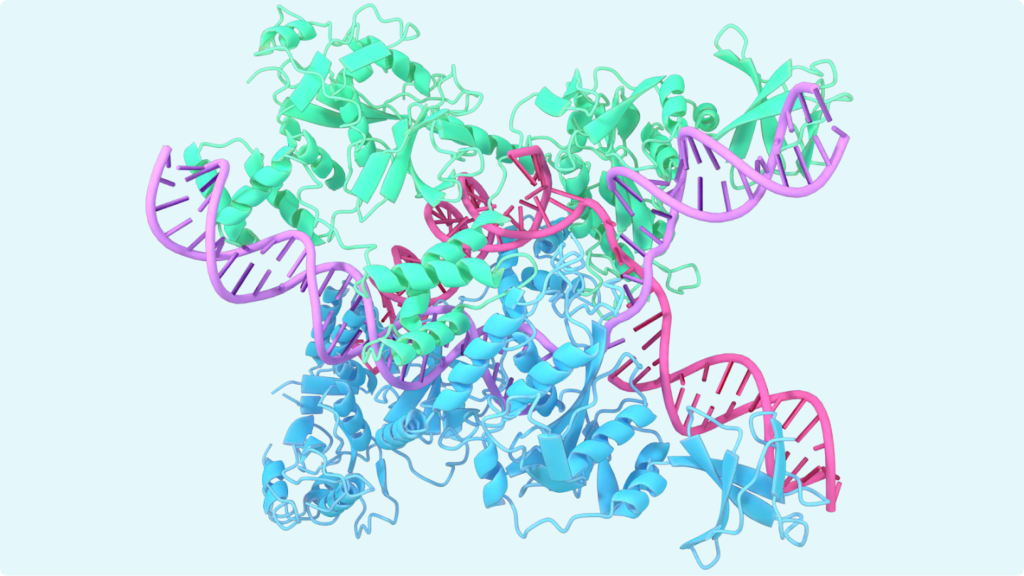
How does AlphaFold Work?
- Database Search: It takes an input of an amino acid sequence. It then compares to find similar proteins in databases to build a multiple sequence alignment.
- Evoformer Architecture: A neural network processes evolutionary and pairwise information to predict distances and orientations between amino acids. It then folds the amino acid chain into a 3D structure.
Resources:
https://blast.ncbi.nlm.nih.gov/Blast.cgi
https://arep.med.harvard.edu/seqanal/blast.html